がん遺伝子c-Mycについて
◉ 遺伝子過剰発現とがん遺伝子中毒
がんは遺伝子の変異や発現異常によって起こる病気です。「がん遺伝子の活性化」や「がん抑制遺伝子の不活性化」などによって、細胞の増殖や生存の制御に異常が生じて、がん細胞が発生します。
発がんは一つの遺伝子の異常によって起こる単純なプロセスではなく、複数の遺伝子の変異が積み重なって起こります。 発がんが極めて複雑な要因によりもたらされるにもかかわらず、ある一つの遺伝子を正常化することによりがん細胞の増殖が劇的に抑制されることが示されています。 つまり、これらのがん細胞は悪性形質を維持するために、ある特定の遺伝子の持続的な活性化に強く依存していることを示唆しています。
これを2002年にWeinsteinは「oncogene addiction」という言葉で定義しています。 Addictionは「Drug addiction(薬物中毒、薬物依存)」や「alcohol addiction(アルコール中毒、アルコール依存)」と同じで「中毒」や「依存」という意味です。 つまり、「oncogene addiction」は「がん細胞が,増殖・進展するうえで特定のシグナル伝達経路に強く依存していること」を意味し、「がん遺伝子中毒」や「がん遺伝子依存」と訳されています。
例えば、乳がんにおけるHER2や神経芽細胞腫のN-Mycの過剰発現やなどがあげられます。 がん細胞が中毒(依存)に陥っている遺伝子異常はがん細胞のアキレス腱となる可能性を秘めています。つまり、このような遺伝子異常は治療標的として創薬につながる可能性の高い遺伝子と言えます。
正常な細胞では、様々な遺伝子が「必要なときに必要な量」発現することによって正常な働きを実行できます。遺伝子が通常の量よりも過剰に発現してしまうことを「過剰発現」といいます。
遺伝子が過剰発現すると、細胞機能は様々な変調を来します。 がん細胞ではがん遺伝子の過剰発現が多く観察されます。過剰発現している遺伝子にがん細胞は依存性が高い状態にあると言えます。したがって、過剰発現しているがん遺伝子は治療のターゲットになります。
例えば、c-Myc遺伝子が過剰発現しているがん細胞でc-Myc遺伝子の発現を押さえるとがん細胞が死んでしまいます。これはがん細胞の生存がc-Myc遺伝子に強く依存しているということを表しています。 多くのがん細胞でc-Myc遺伝子の過剰発現が起こっており、c-Mycはがん治療の重要なターゲットになっています。
現時点で、c-Mycの働き自体を直接阻害する薬はありませんが、c-Mycの発現レベルを阻害する方法は幾つかあります。c-Myc遺伝子の発現を抑制する治療法はがん治療に有効です。
実際に、MYCの不活性化はヒトがんの治療に有効であることが明らかになっています。 c-Mycの不活性化はリンパ腫、白血病、骨肉腫、肝臓がん、扁平上皮癌、すい臓がんなど多くのがん種に対して、増殖抑制、最終分化誘導、アポトーシス誘導、血管新生抑制など多彩な機序で抗腫瘍効果を示すことが報告されています。
◉ c-Mycタンパク質は転写因子
c-Mycはバーキットリンパ腫の原因遺伝子として見つかりました。 バーキットリンパ腫(Burkitt lymphoma)は、主に小児に生じる悪性リンパ腫の一種で、リンパ球の中のB細胞から発生するリンパ腫です。中央アフリカで風土病となっており、米国では小児リンパ腫の約30%を占めます。 病名は、外科医のデニス・バーキット(Denis Parsons Burkitt)が1958年にアフリカで小児に発症する腫瘍を報告したことに由来します。
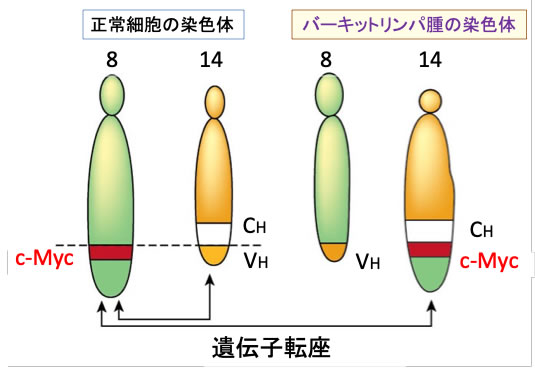
c-Myc遺伝子が染色体転座によりイムノグロブリン重鎖遺伝子のエンハンサーにより制御されるようになったことでc-MycがB細胞において過剰発現し、リンパ腫を引き起こします(下図)。
図:正常ではc-Myc遺伝子は8番染色体に存在し、イムノグロブリン(抗体)の重鎖(heavy chains)遺伝子は14番染色体に存在する。バーキットリンパ腫細胞では、c-Myc遺伝子がイムノグロブリン重鎖遺伝子のエンハンサー(CH)の下流に転座している。その結果、c-Myc遺伝子が過剰発現して、B細胞が腫瘍化する。
c-Mycのcはcellular(細胞の)のcであり、トリ白血病ウイルスがもつv-Myc遺伝子の細胞性のカウンターパートして同定された遺伝子です。バーキットリンパ腫細胞の原因遺伝子として発見されたあと、多くの種類のがんにおいて, c-Mycタンパク質の安定性が向上する変異や遺伝子増幅による発現上昇などが認められています。
c-Mycは、腫瘍化に深くかかわる遺伝子として古くから知られており、さらに幹細胞維持にもかかわっていることや、iPS細胞誘導のための初期化因子の一つであることが示されています。
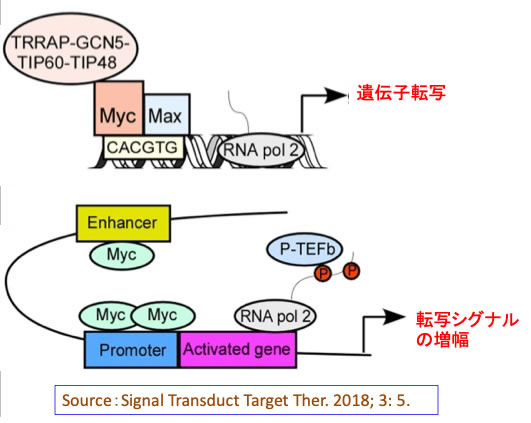
c-Mycは細胞増殖や嫌気的代謝を促進する因子としてがん細胞の特質に大いにかかわっている遺伝子であり、その遺伝子産物であるc-Mycタンパク質はパートナー因子であるMax(MYC-associated protein X)と相互作用することで転写因子として機能します。 さらに、c-Mycタンパク質は遺伝子のプロモーターやエンハンサー領域に集まって、転写シグナルを増幅する働きもあります(下図)。
図:(上)MycはMaxと二量体を形成し、E-box(Enhancer box)と呼ばれるCACGTG配列に結合し、クロマチン構造を制御するタンパク質(GCN5, TIP60, TIP48, TRRAPなど)をリクルートし遺伝子転写を活性化する。GCN5 とTIP60はヒストンアセチル基転移酵素、TIP48はATP結合タンパク質、TRRAPは形質転換/転写ドメイン関連タンパク質。
(下)Mycタンパク質はE-boxのみでなく、遺伝子のプロモーターやエンハンサー領域に集まって、転写シグナルを増幅する働きがある 。
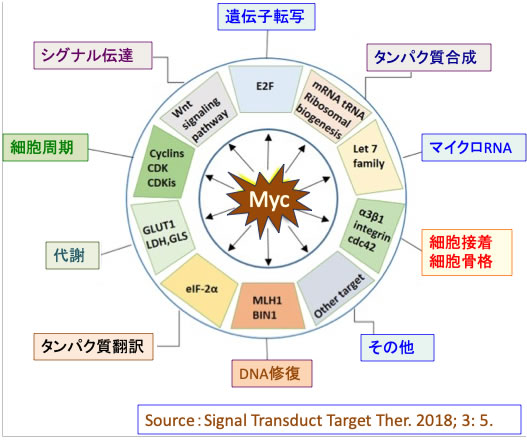
c-Mycは細胞内の様々な機能を制御しています。タンパク質をコードしている遺伝子だけでなく、タンパク質をコードしていない遺伝子の制御にも関わっており、細胞周期、タンパク質合成、細胞接着、代謝、シグナル伝達、遺伝子転写、タンパク質翻訳、など多くの細胞機能を制御しています。(下図)
図:Mycは様々な遺伝子の転写を制御し、多彩な細胞機能の制御に関与している。
◉ Wnt/β-カテニン経路の活性化はc-Mycの発現を亢進する
Wntシグナルは種を超えて広く保存されたシグナル伝達経路で、遺伝子発現、細胞増殖、細胞運動、細胞極性などを調節することで、発生や幹細胞の維持、発がんなどに深く関与することが知られています。 特にβカテニンを介するWnt/β-カテニン・シグナル伝達系は多くのがん細胞で異常を起こしており、がん治療の重要なターゲットになっています。
βカテニンは781個のアミノ酸からなる92kDaのタンパク質で、細胞間接着と遺伝子発現調節の2つの働きを持っています。
βカテニンの大部分は細胞間接着結合部位に局在し, 膜貫通型の接着タンパクであるE-カドヘリン(E-Cadherin)と会合体を作っています。このような細胞膜の接着部位のβカテニンはE-カドヘリンとアクチン細胞骨格との連結を助けています。
E-カドヘリンと会合していないβカテニンはすべて細胞質で複数のタンパク質からなる大型の分解複合体により分解されています。 しかし、Wnt(ウィント)という分子量約4万の分泌性糖タンパク質が受容体に結合すると、細胞質におけるβ-カテニンの分解が阻止されて細胞質に蓄積し、核内に移行した後, 転写因子のTcf/Lef(T cell factor/ Lymphoid enhancer factor)と複合体を形成し、Tcf/Lefの転写活性を亢進します。
つまり、β-カテニンはTcf/Lefの転写活性化補助因子として機能し、Tcf/Lefの標的遺伝子の転写を誘導します。このシグナル伝達系をWnt/β-カテニン経路と言い、この経路のターゲット遺伝子にc-Mycが含まれます。(下図)
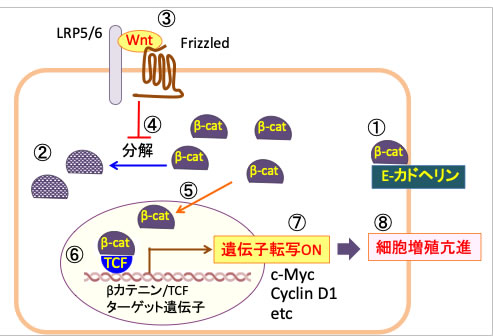
図:β-カテニンは細胞間接着結合部分に局在し, 膜貫通型の接着タンパクであるE-カドヘリンに結合し、カドヘリンとアクチン細胞骨格との連結を助けている(①)。E-カドヘリンと会合していないβ-カテニンはリン酸化され、ユビキチン化を受けて最終的にプロテアソームで分解される(②)。Wntは細胞膜上のFrizzled(7回膜貫通型受容体)と共役受容体である1回膜貫通型LRP5/6(Low-density lipoprotein receptor-related protein5/6)に結合する(③)。Wntが受容体に結合するとβ-カテニンのリン酸化が抑制され、β-カテニンの分解が阻止される(④)。β-カテニンは細胞質に蓄積し核内に移行し(⑤)、転写因子のTCF(T cell factor)と複合体を形成する(⑥)。βカテニンにより活性化される遺伝子群にはc-Mycや cyclinD1など細胞の増殖を促進する因子が含まれ(⑦)、その結果、細胞の増殖が亢進する(⑧)。
β-カテニンは細胞膜近傍か細胞質・核のどちらかに局在し, 特に核にあるときは一連の遺伝子発現に影響を与えると考えられています。 Wnt/β-カテニン・シグナル伝達系により活性化される遺伝子群にはc-Myc、 c-Jun、 cyclinD1など細胞の増殖や転移を促進する因子が含まれます。つまり、Wnt/β-カテニン・シグナル伝達系が活性化されると、がん細胞の増殖や転移が促進されることになります。
がん細胞ではWnt/β-カテニン経路の異常が高頻度で認められます。Wnt/β-カテニン経路は極めて複雑で、まだ不明な点も多くあります。簡単にまとめると、次のようになります。
1)Wntは分子量約4万の細胞外分泌糖タンパク質で、種を超えて保存されており、初期発生における体軸の決定や器官形成を制御しています。これまでに哺乳類において19種類のWnt が同定されています。
2)Wnt はFrizzledやlow-density lipoprotein receptorrelated protein(LRP)5/6の受容体を介して細胞内にシグルを伝達し、多様な細胞機能を制御しています。Frizzledは7回膜貫通型受容体でLRP5/6はFrizzledの共役受容体として機能します。
3)Wnt の非存在下では細胞質内のβ-カテニンのタンパク質量は低く保たれています。これはGSK-3がβ-カテニンをリン酸化し、リン酸化された-カテニンはユビキチン化を受け、最終的にはプロテアソームで分解されるためです。
4)Wnt が分泌されて細胞膜上のFrizzled と共役受容体であるLRP5/6に結合すると,そのシグナルは細胞内へと伝達され、GSK-3依存性のβ-カテニンのリン酸化を抑制し、低リン酸化状態となったβ-カテニンはプロテアソームによる分解から免れ、細胞質内に蓄積します。
5)細胞内に蓄積したβ-カテニンは核内に移行し、転写因子Tcf/Lef と複合体を形成して標的遺伝子の発現を促進することによって、種々の細胞機能を制御しています。Tcf/LefはT-cell factor/lymphoid enhancer factorの略です。
6)Tcf/Lefの標的遺伝子は100種類以上に及び、細胞の増殖、分化、運動、幹細胞多能性維持などの制御に関わっています。c-mycやcyclin D1などの発現を亢進して細胞増殖を促進します。(下図参照)
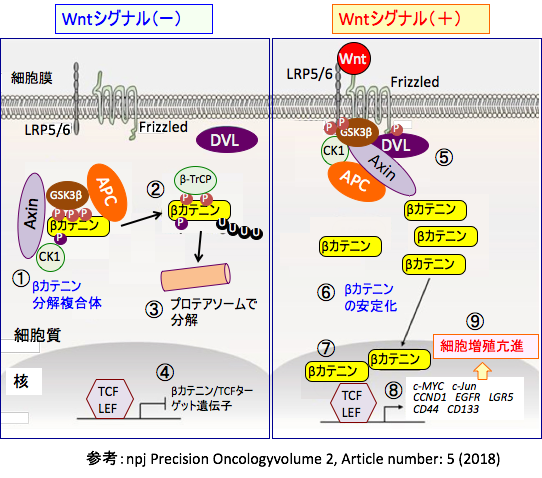
図:(左)Wntのシグナルが無い状況では、β-カテニンは細胞質内で分解複合体によってリン酸化され(①)、ユビキチン(U)が結合し(②)プロテアソームで分解されている(③)。その結果、Tcf/Lef(T-cell factor/lymphoid enhancer factor)による遺伝子発現が阻止されている(④)。
(右)Wntが受容体のFrizzledとLRP5/6に結合してWntシグナルが活性化されると、分化複合体が不活性化され、β-カテニンのリン酸化が阻止されて(⑤)β-カテニンは分解されなくなり(⑥)、細胞質内で増加し核内に移行して転写因子のTCFに結合し(⑦)、β-カテニン/TCFのターゲット遺伝子の転写を活性化して(⑧)、細胞の増殖を亢進する(⑨)。 βカテニン分解複合体は、AXIN、APC(adenomatous polyposis coli) 、セリン・スレオニンキナーゼのGSK3β(glycogen synthase kinase-3)、CK1α (casein kinase 1α)から構成され、GSK3βとCK1αがβカテニンをリン酸化する。βカテニンがリン酸化されるとβ‑TrCP E3 linker によってユビキチンが結合して、プロテアソームで分解される。 (参考:参考:npj Precision Oncologyvolume 2, Article number: 5 (2018) )
◉ 駆虫薬のメベンダゾールはWnt/βカテニンによる遺伝子発現を阻害する
がん細胞では様々なメカニズムでWnt/β-カテニン・シグナル伝達系が活性化され、核内でのβ-カテニンの量が増加し、その結果、TCf/Lefの転写因子活性が亢進して、がん細胞の増殖を促進しています。したがって、Wnt/β-カテニン・シグナル伝達系の阻害はがん治療のターゲットとして重要視されています。
駆虫薬のメベンダゾールがWnt/β-カテニン・シグナル伝達系の最下流の遺伝子発現レベルで阻害作用を示すことが報告されています。以下のような報告があります。Comprehensive Modeling and Discovery of Mebendazole as a Novel TRAF2- and NCK-interacting Kinase Inhibitor.(包括的モデリングと新規TRAF2およびNCK相互作用キナーゼ阻害剤としてのメベンダゾールの発見)Sci Rep. 2016 Sep 21;6:33534. doi: 10.1038/srep33534.
前述のように、TRAF2およびNCK相互作用キナーゼ(TRAF2- and NCK-interacting kinase :TNIK)は、Wntシグナル系が活性化した結腸直腸がんの重要なターゲットの1つです。この研究では、TNIK阻害剤を探索するために1,448種類の FDA(米国食品医薬品局)承認の小分子薬物のスクリーニングに行いました。 コンピュータを使った構造解析や結合活性の解析の結果、FDA承認の駆虫薬であるメベンダゾールは、解離定数Kd =〜1μMでTNIKキナーゼ活性を選択的に阻害することが発見されました。
がん治療薬の開発では、培養がん細胞(in vitro)や移植腫瘍などを使った動物実験(in vivo)で抗がん活性や安全性や薬物動態が検討されます。 さらに最近は、薬剤の候補物質がデータベース化され、細胞の受容体やシグナル伝達物質の構造のデータベースや、抗がん剤による遺伝子発現パターンのデータベースなど様々な情報をコンピューターを使って探索する方法(in silico)もあります。 「in silico」という用語は,「コンピュータ(シリコンチップ)の中で」の意味で、in vitro(試験管内で)やin vivo(生体内で)に対応して作られた用語で、コンピューターを駆使した研究です。
TNIK(TRAF-2 and NCK-interacting kinase)はセリン・スレオニンキナーゼで、このキナーゼ活性(タンパク質をリン酸化する活性)は結腸直腸がんの増殖活性の維持に必須であることが報告されています。 Wnt/βカテニン経路の最終段階であるβカテニンとTCFの相互作用において、TNIKはTCFのセリン154をリン酸化します。このリン酸化がβカテニン/TCFの遺伝子転写活性に必要だと言うことです。
したがって、TNIKの阻害剤は大腸がんのようにWnt/βカテニンシグナル伝達系が亢進したがんの治療に有効と考えられており、多くの製薬会社が開発しています。まだ臨床的に使用できるものはありませんが、何十年も前から多くの国で使用されている駆虫薬のメベンダゾールが、TNIKの阻害剤としてかなり有望だという報告です。
以下のような報告もあります。メベンダゾールの抗腫瘍効果にc-Mycが関与しているという報告です。
Mebendazole induces apoptosis via C-MYC inactivation in malignant ascites cell line (AGP01).(メベンダゾールは悪性腹水細胞株AGP01においてC‐MYC不活性化を介してアポトーシスを誘導する)Toxicol In Vitro. 2019 Jun 14;60:305-312.
この論文では、メベンダゾールは悪性腹水細胞のAGP01細胞のDNA損傷を有意に増加させましたが、正常ヒトリンパ球に対してはDNA損傷を引き起こしませんでした。 メベンダゾールは、0.5μMおよび1.0 μMの濃度で、それぞれG0/G1 および G2/M期において顕著な細胞周期停止を引き起こし、そしてより高濃度で有意にアポトーシスを誘導しました。
さらに、メベンダゾールは、AGP01細胞におけるc-MYC mRNA とc-MYCタンパク質発現を減少させました。 つまり、メベンダゾールががん細胞に細胞死(アポトーシス)を誘導する経路の1つとしてc-MYC遺伝子の関与を示唆しています。 以下の報告もメベンダゾールの抗腫瘍効果にc-Mycの関与を報告しています。
Anthelmintic mebendazole enhances cisplatin's effect on suppressing cell proliferation and promotes differentiation of head and neck squamous cell carcinoma (HNSCC).(駆虫薬メベンダゾールはシスプラチンの細胞増殖抑制効果を高め、頭頸部扁平上皮がんの分化を促進する)Oncotarget. 2017 Feb 21;8(8):12968-12982.
この論文では、2種類のヒト頭頸部扁平上皮がん細胞株のCAL27およびSCC15を使用して、メベンダゾールがヒト頭頸部扁平上皮がん細胞においてシスプラチンよりも強力な抗増殖活性を発揮することを示しています。 メベンダゾールは頭頸部扁平上皮がん細胞の細胞増殖、細胞周期進行および細胞移動を効果的に阻害し、そしてアポトーシスを誘導しました。 メカニズム的には、メベンダゾールはELK1/SRF、AP1、STAT1/2、MYC/MAXを含むがん関連経路を制御しました。 メベンダゾールはまた、頭頸部扁平上皮がん細胞の細胞増殖の抑制およびアポトーシスの誘導においてシスプラチンと相乗的に作用しました。 さらに、メベンダゾールは、CAL27細胞の最終分化およびCAL27細胞の異種移植腫瘍の角質化を促進しました。
これらの結果は、メベンダゾールが頭頸部扁平上皮がん細胞の分化を促進しながら増殖を阻害することによってその抗がん活性を発揮し得ることを示しています。そのメカニズムとしてc-Mycの阻害の関与を指摘しています。
駆虫薬のメベンダゾールは、頭頸部扁平上皮がん治療においてシスプラチンなどの他の化学療法薬と組み合わせて使用される安全かつ有効な薬剤として再利用され得ると指摘しています。 がん治療以外で使用されている医薬品の抗がん作用をスクリーニングして、がん治療に利用できる薬を見つけようという医薬品再利用においてメベンダゾールは有力な候補として研究されています。以下のような報告もあります。
Repositioning of the anthelmintic drug mebendazole for the treatment for colon cancer.(寄生虫治療薬メベンダゾールの大腸がん治療薬としての再開発)J Cancer Res Clin Oncol. 139(12): 2133-40, 2013年
この論文では、2種類の大腸がん細胞株を用い、臨床的に使用されている1600種類の医薬品を含む多数の物質をスクリニーングしました。 10μMの濃度で細胞生存率を40%以下に減らす細胞傷害活性を示す物質が68種類スクリーニングされ、その一つが寄生虫治療薬のベンゾイミダゾール(Benzimidazole)系の薬物でした。このうちメベンダゾール(Mebendazole)とアルベンダゾール(Albendazole)が臨床で使用されています。 60種類のがん細胞株を用いた薬剤感受性のデータベースとの比較から、メベンダゾールとアルベンダゾールの類似性は低く、作用機序が異なることが示唆されました。メベンダゾールはBCR-ABLやBRAFを含む幾つかのプロテインキナーゼと相互作用を示しましたが、アルベンダゾールにはそのような作用は認めませんでした。 メベンダゾールはNCA60パネルの大腸がん細胞株の80%に対して抗腫瘍活性を示しました。さらに3種類の大腸がん細胞と3種類の非がん細胞での検討から、メベンダゾールの大腸がんに対する選択性が確認されました。
この論文の結論は『メベンダゾールは大腸がん治療薬として再開発する価値がある』と記載されています。メベンダゾールはがんの代替医療ではかなり有名です。 駆虫薬として入手は容易で、副作用も極めて少ない薬です。がんに対する有効性も証明されています。
メベンダゾールの消化管からの吸収が低いのが欠点ですが、油の多い食事の後に服用すると吸収率を高めることができますので、ケトン食との併用は有効です。 また、メベンダゾールを分解する薬物代謝酵素を阻害するグレープフルーツや胃薬のシメチジンを併用すると、メベンダゾールの血中濃度を高めることができます。
◉ AMPKの活性化はmTORC1を阻害してc-Myc発現を抑制する
AMP活性化プロテインキナーゼ (AMPK)はグルコース欠乏や低酸素などにより細胞内ATP量が減少すると、AMP/ATP 比の増加に伴いAMPKが活性化されます。AMPKはmTORC1活性を抑制して、異化作用の亢進や細胞成長の停止をもたらし、エネルギー消費の抑制(同化抑制)とエネルギー産生の亢進(異化促進)へと細胞の代謝をシフトさせます。(下図)
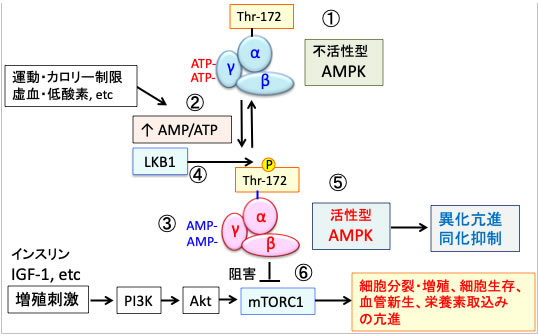
図:AMPKは触媒作用を持つαサブユニットと、調節作用を持つβサブユットとγサブユニットから構成されるヘテロ三量体として存在する(①)。運動やカロリー制限や虚血や低酸素などによってATPが減少してAMP/ATP比が上昇すると(②)、γサブユニットに結合していたATPがAMPに置換する(③)。これによってAMPKの構造変化が起こると、LKB1というリン酸化酵素の親和性が高まり、αサブユニットのスレオニン172がリン酸化されると、さらにAMPKの活性が高まる(④)。活性化したAMPKは異化を亢進してエネルギー産生を亢進し、物質合成(同化)を抑制するように代謝をシフトする(⑤)。活性化したAMPKはmTORC1(哺乳類ラパマイシン標的タンパク質複合体1)を阻害し(⑥)、がん細胞の増殖抑制や、抗老化や寿命延長の効果を引き起こす。
AMPKは運動やカロリー制限の他、メトホルミンやビタミンD3で活性化できます。
メトホルミンはミトコンドリアの呼吸鎖を阻害してATP産生を低下させる機序とLKB1を活性化する両方の機序でAMPKを活性化します。
ビタミンD3は細胞内のフリーのカルシウムを増加させ、カルモジュリンキナーゼキナーゼβ (CaMKKβ)を活性化させてAMPK活性を亢進します。つまり、異なる機序でAMPKを活性化するので、相乗効果が期待できます。(下図)。
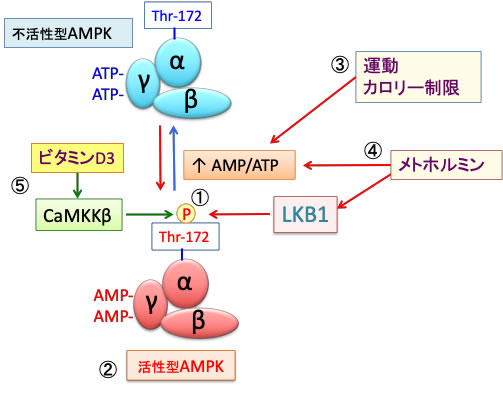
図:AMPKの172番目のスレオニン(Thr-172)がLKB1でリン酸化されると(①)、AMPKは最大に活性化される(②)。運動やカロリー制限はATPが減少してAMP/ATP比を上昇してAMPKを活性化する(③)。メトホルミンはミトコンドリアの呼吸鎖を阻害してATP産生を低下させる機序とLKB1を活性化する両方の機序でAMPKを活性化する(④)。ビタミンD3は細胞内のフリーのカルシウムを増加させ、カルモジュリンキナーゼキナーゼβ (CaMKKβ)を活性化させてAMPK活性を亢進する(⑤)。
AMPKの活性化はmTORC1活性を阻害するので、c-Mycを阻害する可能性があります。以下のような報告があります。
Metformin targets c-MYC oncogene to prevent prostate cancer(メトホルミンはc-MYC癌遺伝子を標的にして前立腺がんを予防する)Carcinogenesis. 2013 Dec;34(12):2823-32.
【要旨】
疫学研究は、糖尿病薬メトホルミンを投与されている患者は前立腺がんリスクが低く、予後が良好であることを示しており、メトホルミンが前立腺がんに対して抗腫瘍効果を有する可能性があることを示唆している。 メトホルミンが前立腺がんの発生と進行を妨げる化学予防剤として作用するメカニズムは不明である。c-MYCがん遺伝子の増幅は、前立腺上皮細胞の発がん過程と前立腺がんの増殖に重要な役割を果たしている。 この研究の目的は、c-mycの発現と前立腺がんの進行に対するメトホルミンの効果を検討することである。 我々の実験は以下の結果が得られた。
(i)Myc高発現マウス(Hi-Mycマウス)は、人間の前立腺がんの進行に非常に類似した前立腺腫瘍を発症する。この動物モデルでメトホルミンは前立腺上皮内腫瘍(前立腺の前がん病変)および前立腺がんの発症を減弱させた。
(ii)in vivoおよびin vitroの実験系で、メトホルミンはc-mycタンパク質レベルを少なくとも50%減少させた。
(iii)メトホルミンは、正常な前立腺上皮細胞の増殖に影響を与えることなく、前立腺がん細胞の細胞周期の停止とアポトーシス誘導により、前立腺がん細胞の増殖を選択的に阻害した。
(iv)Hi-Mycマウスにおけるメトホルミンによる前立腺腫瘍の抑制は、前立腺におけるアンドロゲン受容体および増殖マーカーKi-67のレベルの低下と関連していた。 これらの実験結果は、メトホルミンは前立腺上皮細胞におけるc-myc発現を抑制することによって、前立腺がんの発生と進展を予防する化学予防剤として作用することを示唆している。
まとめ:古い糖尿病治療薬であるメトホルミンは、古くから知られているがん遺伝子であるc-MYCを減少させることにより、前立腺上皮内腫瘍ががん病変に変化するのを阻害する可能性がある。この研究では、前立腺がんにおけるメトホルミンの化学的予防効果と、in vitroおよびin vivoでのc-MYCとの関連を明らかにしている。
以下のような報告があります。AMPK/mTORシグナル伝達経路の阻害作用においてビタミンD3とメトホルミンは相乗効果があるという報告です。
Vitamin D3 potentiates the growth inhibitory effects of metformin in DU145 human prostate cancer cells mediated by AMPK/mTOR signalling pathway(ビタミンD3 は、AMPK/mTOR シグナル伝達経路を介した DU145 ヒト前立腺がん細胞におけるメトホルミンの増殖抑制効果を増強する)Clin Exp Pharmacol Physiol. 2015 Jun;42(6):711-7
【要旨】
メトホルミンとビタミンD3 は両方とも、ヒト前立腺がん細胞を含む多数のがん細胞株で強力な抗増殖作用を示す。ここでは、2 つの薬剤の組み合わせが、DU145 ヒト前立腺がん細胞の増殖に対して、いずれかの薬剤単独よりもはるかに強い増殖抑制効果を与えることを示した。 メトホルミンとビタミン D3 は、培養 DU145 細胞の細胞増殖とアポトーシス誘導に対して相乗効果を示した。 抗腫瘍作用のメカニズムとして、G1/S期での細胞周期の停止、AMPKのリン酸化による活性化とそれに続く下流のmTORシグナル伝達経路の阻害、c-Myc発現の抑制、および抗アポトーシスタンパク質p-Bcl-2のレベルの低下の関与が示された。結論として、メトホルミンとビタミン D3 は相乗的に DU145 細胞の増殖を阻害し、アンドロゲン非依存性前立腺癌の治療に対する有望な臨床治療戦略を示している
メトホルミンとビタミンD3は両方ともAMP活性化プロテインキナーゼを活性化しますが、そのメカニズムは異なります。従って、AMPK活性化において相乗効果が期待できます。
駆虫薬のイベルメクチンはAkt/mTORC1経路を阻害します。さらにWnt/βカテニン経路を阻害する作用もあります。したがって、イベルメクチンとメトホルミンとビタミンD3は相乗効果が期待できます。
◉ DHA/EPAはAMPKを活性化してmTORC1を阻害する
ポイツ・ジェガース症候群(Peutz-Jeghers syndrome)という遺伝性疾患があります。食道を除く消化管に多数のポリープを生じ、口唇、口腔粘膜、手掌、足底に特有な色素斑をみとめます。 ポリープの分布は小腸に多く、大腸には少なく、小腸ポリープが腸重積症の原因となり、イレウスや腹痛をきっかけに診断されることがしばしばあります。長期的にみると、この病気はがんの危険性が高く、特に大腸がんの発生が多くみられます。肺や卵巣や子宮など他の臓器のがんも認められます。 10万人に一人くらいの発症率で、常染色体性優性の遺伝形式をとり、原因遺伝子は19番染色体短腕にあるLKB1遺伝子です。
LKB1はセリン・スレオニンキナーゼで、AMP活性化プロテインキナーゼ(AMPK)をリン酸化して活性化します。 DHA(ドコサヘキサエン酸)やEPA(エイコサペンタエン酸)のようなオメガ3系多価不飽和脂肪酸がLKB1を活性化して、解糖系酵素とmTORシグナル伝達系を阻害するという報告があります。以下のような論文があります。
Omega-3 polyunsaturated fatty acid promotes the inhibition of glycolytic enzymes and mTOR signaling by regulating the tumor suppressor LKB1.(オメガ3系多価不飽和脂肪酸はがん抑制遺伝子のLKB1の制御を介して、解糖系酵素とmTORシグナル伝達系の阻害を促進する)Cancer Biol Ther. 2013 Nov 1; 14(11): 1050–1058.
【要旨】
オメガ3系多価不飽和脂肪酸は、炎症性疾患や心血管系疾患や悪性腫瘍の治療に有効な脂質として知られている。食事からのオメガ3系多価不飽和脂肪酸の摂取量の多い人は、がんを含めて代謝系疾患の発症頻度が低いことが報告されている。 これらの疾患の予防や治療においてオメガ3系多価不飽和脂肪酸の有効性は良く認識されているが、その作用機序については十分に明らかになっていない。 この研究では、オメガ3系多価不飽和脂肪酸のドコサヘキサエン酸(DHA)ががん抑制遺伝子のLKB1の作用を亢進することを明らかにした。
LKB1遺伝子を発現している細胞にDHAを投与すると、LKB1活性が亢進し、AMP活性化プロテインキナーゼ(AMPK)のリン酸化を引き起こし、mTORシグナル伝達系を阻害した。
乳がん細胞MCF-7細胞をsiRNA(干渉RNA)でLKB1発現を阻止すると、DHAのAMPKの活性化とmTORシグナル伝達系阻害作用は見られなかった。 さらに、LKB1遺伝子を発現している細胞では、DHA投与によって細胞内代謝系が変化しATP量は減少した。
さらに重要な点は、LKB1を発現している上皮性細胞において、DHA投与によって細胞の解糖系酵素の発現が低下して解糖の活性が低下した。 その結果、細胞機能においては、これらの細胞の移動能が低下した。
全体的に、今回の研究結果は、オメガ3系多価不飽和脂肪酸がLKB1活性を亢進し、細胞の代謝を制御していることを明らかにした最初の発見である。
この論文の研究結果は、がん抑制遺伝子のLKB1の活性化とAMPKの活性化を介して、がん細胞のワールブルグ効果の是正にDHAやEPAは役立つことを示唆しています。 この論文では、DHA/EPAが、がん抑制遺伝子のLKB1の活性を高めて、AMPKの活性を高め、解糖系酵素(ヘキソキナーゼ-2や乳酸脱水素酵素)の発現を抑制し、mTORシグナル伝達系を抑制して、抗がん作用を示すという新しいメカニズムが提唱されています。この作用はがん細胞のワールブルグ効果を是正するという作用機序です。
慢性炎症は老化と発がん過程を促進します。DHA/EPAは抗炎症作用や細胞保護作用など様々なメカニズムによって老化と発がんを抑制する効果を発揮します。
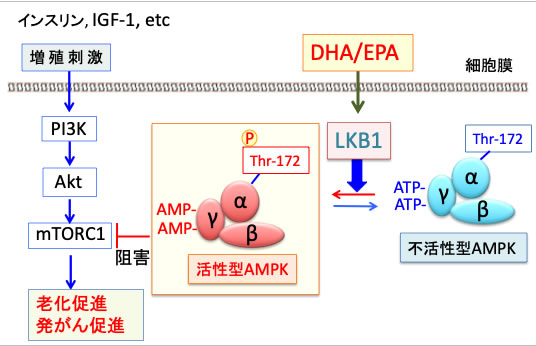
図:インスリンやインスリン様成長因子-1(IGF-1)などの増殖刺激はPI3K/Akt/mTORC1経路を活性化して老化と発がんを促進する作用がある。AMP活性化プロテインキナーゼ(AMPK)はα, β, γの3つのサブユニットからなるヘテロ三量体で、ATPが減少してAMP/ATP比が上昇すると、γサブユニットに結合していたATPがAMPに置換する。これによってAMPKの構造変化が起こると、LKB1というリン酸化酵素の親和性が高まり、αサブユニットのスレオニン172がLKB1によってリン酸化されると、さらにAMPKの活性が高まる。活性化したAMPKはmTORC1(哺乳類ラパマイシン標的タンパク質複合体1)を阻害し、がん細胞の増殖を抑制する。オメガ3系多価不飽和脂肪酸のDHA(ドコサヘキサエン酸)とEPA(エコサペンタエン酸)はLKB1の活性を亢進してAMPKを活性化し、mTORC1を抑制することが報告されている。すなわち、DHA/EPAは老化予防とがん予防効果を発揮する。
◉ ドコサヘキサエン酸はc-Mycタンパク質の分解を促進する
以下のような報告があります。中国医科大学(China Medical University)からの報告です。
Docosahexaenoic Acid Inhibits Cell Proliferation through a Suppression of c-Myc Protein in Pancreatic Ductal Adenocarcinoma Cells(ドコサヘキサエン酸は、膵管腺がん細胞におけるc-Mycタンパク質の抑制により細胞増殖を阻害する)Antioxidants (Basel). 2021 Oct 28;10(11):1721.
【要旨】
生存シグナル伝達経路の異常な活性化を阻害することによる膵臓がんの治療は、かなりの注目を集めている。ヒト膵管腺がん細胞におけるドコサヘキサエン酸(DHA)の細胞増殖に対する作用を検討した。 我々の実験結果は、DHA がヒト膵管腺がん細胞の細胞周期停止の誘導を介して用量依存的に細胞増殖を阻害することを示した。
DHAは、リン酸化 Rb (p-Rb)、サイクリン D1、サイクリン E、サイクリン A、E2F1、および c-Myc タンパク質の発現を抑制した。 STAT3 シグナル伝達経路の活性化を阻害すると、Ca2+/カルモジュリン依存性プロテインキナーゼII(CaMKII)が不活性化され、c-Myc (T58) タンパク質のリン酸化が増加し、c-Myc タンパク質の発現量が減少した。
DHA の処理は、EGFR、STAT3、および CAMKII タンパク質のリン酸化レベルの低下を通じて、細胞の生存を効果的に阻害した。
作用機序は、c-Myc (T58) のリン酸化レベルの増加と c-Myc タンパク質の不安定性に関連していた。 DHA は、HPAF-II 細胞の GSSG/GSH 比と酸化ストレスレベルの増加により、細胞の生存を阻害した。 DHA は、HPAF-II 細胞における Bax、c-カスパーゼ 3、および c-PARP タンパク質の発現増加を介して細胞アポトーシスを亢進した。さらに、DHA の処理はヌクレオチド合成を有意に阻害した。
結論として、DHA は 膵管腺がん細胞の増殖を有意に抑制し、したがって抗がん治療薬としての可能性を秘めている可能性がある。
同様の論文は韓国のソウル大学校( Seoul National University)からも報告されています。
RvD1 inhibits TNFα-induced c-Myc expression in normal intestinal epithelial cells and destabilizes hyper-expressed c-Myc in colon cancer cells(RvD1は、正常な腸上皮細胞でTNFα誘導性のc-Myc発現を阻害し、結腸がん細胞で過剰発現したc-Mycを不安定化する)Biochem Biophys Res Commun. 2018 Feb 5;496(2):316-323.
【要旨】
潰瘍性大腸炎やクローン病などの炎症性腸疾患は、持続性炎症によって大腸がんの発生に原因となる。大腸の炎症の収束を促進することは、大腸がん発生の予防のための治療戦略になる。
レゾルビン D1 (RvD1) はドコサヘキサエン酸から生成される内因性脂質メディエーターで、炎症の収束を促進する。 レゾルビンD1の炎症収束を促進する効果は広く研究されているが、大腸炎関連の大腸発がんにおける レゾルビンD1の役割はほとんど不明のままである。この研究では、レゾルビンD1が腫瘍壊死因子-α(TNFα)で刺激された正常な結腸細胞、および結腸がん細胞においてc-Mycの発現を阻害することを発見した。 正常細胞におけるTNF-α誘導性のc-Mycの発現亢進の抑制は、NF-κBシグナル伝達の抑制が関連していた。
特に注目されるのは、レゾルビンD1は、c-Mycタンパク質のユビキチン化とその後のプロテアソーム分解を刺激することにより、HCT 116 ヒト結腸がん細胞で恒常的に過剰発現した c-Myc タンパク質の分解を促進してc-Mycタンパク量を減少させた。さらに、レゾルビンD1 が ALX/FPR2 受容体との直接的な相互作用を介して c-Myc 分解を刺激することを明らかにした。この相互作用は、細胞外シグナル調節キナーゼ(Erk)の活性化の阻害をもたらし、それによって c-Myc のリン酸化依存性安定化を弱めた。
以下の論文は中国の青島大学(Qingdao University)の研究者からの報告です。
Docosahexaenoic acid inhibited the Wnt/β-catenin pathway and suppressed breast cancer cells in vitro and in vivo(ドコサヘキサエン酸は、in vitro および in vivo で Wnt/β-カテニン経路を阻害し、乳がん細胞を抑制する)J Nutr Biochem. 2014 Feb;25(2):104-10.
【要旨】
N-3脂肪酸は、人間の健康に必要な必須脂肪酸であり、抗がん作用を有することが知られている。しかし、マウス乳がんにおける n-3脂肪酸とWnt/β-カテニンシグナル伝達経路との間の関係は、十分に検討されていない。 この研究では、4T1マウス乳がん細胞を用いて、代表的な n-3脂肪酸であるドコサヘキサエン酸 (DHA) のWnt/β-カテニン シグナル伝達経路に対する作用をin vivo および in vitro の実験系で検討た。
In vitro 研究では、4T1 マウス乳がん細胞と MCF-7 ヒト乳がん細胞の両方で 、DHAが細胞増殖を阻害し、G1期での細胞周期の停止を誘導することが示された。 DHAは、4T1マウス乳房細胞におけるβ-カテニンの発現およびT細胞因子/リンパ球増強因子(T cell factor/lymphoid-enhancing factor)の転写因子活性を低下させた。
さらに、DHA は、c-myc やサイクリン D1 などのWnt/βカテニン経路の下流に位置する標的遺伝子の発現を抑制した。
in vivoの実験系で、乳がんを有するBabl/cマウスで治療実験が行われた。マウスに 5% 魚油添加食餌を 30 日間与えると、乳がん細胞の増殖の阻害とアポトーシスの誘導により、4T1 マウス乳がんの成長が大幅に減少することが示された。 動物に 5% 魚油食を与えると、腫瘍組織の β-カテニンの有意な発現抑制が誘導され、アポトーシスが顕著に増加した。さらに、魚油を補給した食事は乳がんの肺転移を減少させた。
これらの観察結果は、DHA が Wnt/β-カテニン シグナル伝達の抑制を介してその抗がん活性を発揮したことを示唆している。したがって、私たちのデータは、乳がんの予防と治療における栄養補助食品としての魚油の有効性を評価するためのさらなる研究の必要性を示している。
次の論文は韓国の忠南大学校(Chungnam National University)からの報告です。
Omega-3-polyunsaturated fatty acids suppress pancreatic cancer cell growth in vitro and in vivo via downregulation of Wnt/Beta-catenin signaling(オメガ-3-多価不飽和脂肪酸は、Wnt/ベータ-カテニンシグナル伝達の抑制を介して、in vitro および in vivo で膵臓がん細胞の増殖を抑制する)
【要旨】
背景/目的: ω3-多価不飽和脂肪酸は、抗がん特性を有することが知られている。しかし、ヒト膵臓がん細胞におけるω3-多価不飽和脂肪酸とWnt/β-カテニンシグナル伝達経路のとの関係は、まだ十分に検討されていない。
方法: ヒト膵臓がん細胞 (SW1990 および PANC-1) を ω3-多価不飽和脂肪酸のドコサヘキサエン酸 (DHA) およびエイコサペンタエン酸 (EPA) に曝露し、ω3-多価不飽和脂肪酸と Wnt/β-カテニン シグナル伝達経路との関係をin vitroで検討した。 ω3 デサチュラーゼ(ω3 desaturases)を発現し、内因的にω3-多価不飽和脂肪酸レベルの上昇をもたらすマウス膵臓がん (PANC02) 細胞を、 fat-1 トランスジェニック マウスに移植された。腫瘍サイズ、Wnt/β-カテニンシグナル伝達分子のレベル、およびアポトーシスレベルを分析して、in vivoでのω3-多価不飽和脂肪酸の影響を調べた。
結果: DHA と EPA は、膵臓がん細胞の細胞増殖を有意に抑制し、細胞死を増加させた。 DHAはまた、β-カテニン発現、T細胞因子/リンパ球増強因子(T cell factor/lymphoid-enhancing factor)レポーター活性を低下させ、β-カテニン分解の前駆体であるβ-カテニン/Axin/GSK-3β複合体形成を誘導した。さらに、天然のWntリガンドのWnt3aは、PANC-1 細胞における DHA 誘導増殖阻害を逆転させた。 免疫組織化学分析は、膵臓がん患者の腫瘍組織におけるβ-カテニンの異常な発現亢進と核染色の増加を示した。しかし、コントロール マウスと比較して、fat-1 トランスジェニックに移植してω3-多価不飽和脂肪酸レベルの上昇をもたらすマウス膵臓がんマウスの腫瘍組織のβ-カテニン レベルは減少し、アポトーシスが有意に増加した。
結論: ω3-多価不飽和脂肪酸は、ヒト膵臓癌の化学予防および治療に有効な治療法である可能性がある。
以上から、DHAはc-Mycタンパク質の分解を促進する機序などでc-Mycの活性を低下させると考えられます。
メベンダゾール、メトホルミン、ビタミンD3、イベルメクチン、ドコサヘキサエン酸はc-Myc遺伝子の転写を異なるメカニズムで阻害するので、相乗効果が期待できます。(下図)
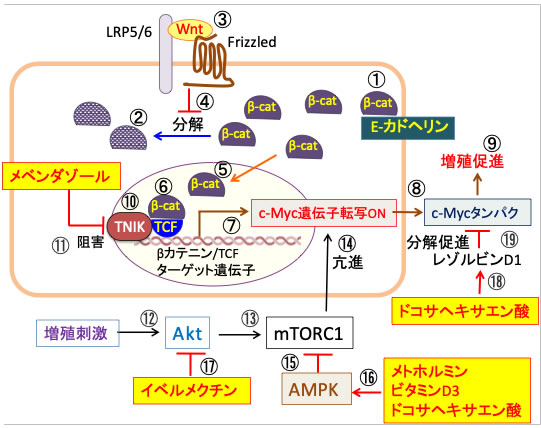
図:β-カテニン(β-cat)は細胞間接着結合部分に局在し, 膜貫通型の接着タンパクであるE-カドヘリンに結合し、カドヘリンとアクチン細胞骨格との連結を助けている(①)。E-カドヘリンと会合していないβ-カテニンはリン酸化され、ユビキチン化を受けて最終的にプロテアソームで分解される(②)。Wntは細胞膜上のFrizzled(7回膜貫通型受容体)と共役受容体である1回膜貫通型LRP5/6(Low-density lipoprotein receptor-related protein5/6)に結合する(③)。Wntが受容体に結合するとβ-カテニンのリン酸化が抑制され、β-カテニンの分解が阻止される(④)。βカテニンは細胞質に蓄積し核内に移行し(⑤)、転写因子のTCF(T cell factor)と複合体を形成し(⑥)、c-Myc遺伝子の転写のスイッチがオンになり(⑦)、c-Mycタンパク質が合成される(⑧)。c-MycはATPと核酸とタンパク質の合成を促進し、細胞増殖を亢進する(⑨)。β-カテニンとTCFの相互作用において、TNIK(TRAF-2 and NCK-interacting kinase)によるTCFのセリン154のリン酸化が必要(⑩)。メベンダゾールはTNIKの活性を阻害して、βカテニン/TCFターゲット遺伝子のc-Mycの発現を阻害する(⑪)。増殖刺激はAktを活性化し(⑫)、AktはmTORC1を活性化する(⑬)。活性化したmTORC1はc-Myc遺伝子の発現を促進する(⑭)。AMP活性化プロテインキナーゼ(AMPK)はmTORC1を阻害する(⑮)。メトホルミンとビタミンD3とドコサヘキサエン酸はAMPKを活性化する(⑯)。イベルメクチンはAkt活性化を阻害する(⑰)。ドコサヘキサエン酸が代謝されて生成するレゾルビンD1(⑱)はc-Mycタンパク質の分解を促進する(⑲)。以上から、メベンダゾール、メトホルミン、ビタミンD3、ドコサヘキサエン酸、イベルメクチンはc-Myc遺伝子の転写を異なるメカニズムで阻害し、相乗効果が期待できる。
◉ BETファミリータンパク質のBRD4がc-Myc遺伝子の転写に関与
c-Mycは転写レベルでの調節や、RNAおよびタンパク質の安定性での調節といったように、様々なレベルでの調節を受けています。
c-Myc mRNAはかなり不安定であり、c-Myc遺伝子が強く転写されても、細胞内でc-Myc mRNAは蓄積しがたく、さらにc-Myc mRNAは,let-7などいくつかのmiRNAによる翻訳の抑制も受けています。
c-Mycタンパク質の58番目のスレオニン残基がGSK-βによってリン酸化を受けると、プロテオソーム分解系によりすみやかに分解されます。 このようにc-Mycタンパク質は、正常細胞では細胞内にあまり多く蓄積しないようにさまざまな調節を受けています。しかし、がん細胞はそういったc-Mycタンパク質の量を制限する機構から回避することで細胞内に比較的多くの量のc-Mycタンパク質の蓄積を達成しています。
その最も典型的な例としては、がん細胞がリン酸化されないように58番目のスレオニン残基を変異させることで、c-MycCタンパク質の安定性を増していることがあげられます。
転写レベルでの調節では、c-Myc遺伝子プロモーター上にBRD4(ブロモドメイン含有タンパク質4)という転写調節因子が結合し、転写を促進していることがわかっており、小分子化合物を用いてBRD4の活性を抑制することでc-Mycの発現を低下させ、がんを治療しようとする試みがなされています。
BETファミリータンパク質は高アセチル化ヒストンへの結合を介して、がん遺伝子や抗アポトーシスタンパク質の発現を促進する作用があります。 ブロモドメインはヒストンのアセチル化リシンを認識し,制御タンパク質を集めてクロマチン構造や遺伝子発現を制御する機能が知られているタンパク質ドメインです。 ブロモドメイン繰り返し配列および特異的末端配列を持つBET(bromodomain and extra-terminal)ファミリータンパク質としてBRD2,BRD3,BRD4,BRDTが知られています。
ヒストンのアセチル化による遺伝子発現の制御には、アセチル化を促進するヒストンアセチル基転移酵素、ヒストンからアセチル基を除去するヒストン脱アセチル化酵素、ヒストンのアセチル化した部分を認識するBETファミリータンパク質の3つが必要です。
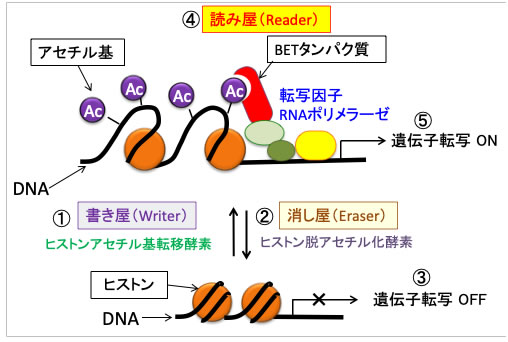
ヒストンアセチル基転移酵素の「書き屋(Writer)」とヒストン脱アセチル化酵素の「消し屋(Eraser)」と、BETファミリータンパク質の「読み屋(Reader)」の3つの役割を担うタンパク質が、ヒストンアセチル化の制御を行ってます。(下図)
図:「書き屋(Writer)」のヒストンアセチル基転移酵素によってヒストンにアセチル基が結合し(①)、「消し屋(Eraser)」のヒストン脱アセチル化酵素によってアセチル基が除去される(②)。ヒストンアセチル化の少ない部分では遺伝子転写は抑制される(③)。「読み屋(Reader)」のBETファミリータンパク質はヒストンのアセチル化リシンと結合して(④)、遺伝子転写を促進する(⑤)。
ヒストンのアセチル化リシンとBETファミリータンパク質のブロモドメインの結合を阻害する薬剤をがん細胞に投与すると、遺伝子発現パターンが正常細胞に近づくことが知られています。
ヒストンとBETファミリータンパク質の結合を阻害する低分子化合物(BET阻害剤)が、がんや炎症性疾患の治療薬として注目されています。臨床試験が行われているBET阻害剤が幾つかあります。 このようなBET阻害剤は、ある種のがん細胞に投与すると腫瘍促進遺伝子の発現を選択的に抑制することから、がん治療薬としての可能性が期待されています。
◉ ニトロキソリンはBETタンパク質を阻害する
抗生物質のニトロキソリンがBETタンパク質を阻害することが報告されています。以下のような論文があります。
Discovery of novel BET inhibitors by drug repurposing of nitroxoline and its analogues.(ニトロキソリンとその類似体の薬物再利用による新規BET阻害剤の発見)Org Biomol Chem. 2017 Nov 15;15(44):9352-9361.
【要旨】
ブロモドメイン含有タンパク質(bromodomain-containing proteins)のBETファミリーは、がん、炎症および心血管疾患を含む多くの疾患の治療に有望な薬物標的であると考えられている。それ故、BET阻害作用のある新規な化合物の開発が注目されている。 安全性および薬物動態が既知の化合物から阻害剤を見出すという薬物再利用戦略は大きな利点を有しており、それ故近年の医薬品開発者の関心を高めている。この薬物再利用戦略を使って、薬物ライブラリーからBRD4特異的阻害作用を有する化合物を探索し、続いてALPHAスクリーニングアッセイ試験を実施した。FDA承認抗生物質であるニトロキソリンは、BRD4(ブロモドメイン含有タンパク質4)の第一ブロモドメインとアセチル化ヒストン4ペプチドとの間の相互作用を50%阻害濃度(IC50)が0.98μMで阻害することを明らかにした。
ニトロキソリンは、非BETブロモドメイン含有タンパク質に対して阻害作用を示さず、良好な選択性で全てのBETファミリーメンバーを阻害した。従ってニトロキソリンは選択的BET阻害剤と呼べる。
ニトロキソリン-BRD4_BD1複合体の結晶構造に基づいて、ニトロキソリンの作用機構およびBET特異性を決定した。 BET関連疾患の1つであるMLL白血病に対するニトロキソリンの抗がん活性はこれまで研究されていなかったので、我々はニトロキソリンがMLL白血病の治療薬として有効かどうかを試験した。ニトロキソリンは細胞周期停止とアポトーシスを誘導することによりMLL白血病細胞の増殖を効果的に抑制した。ニトロキソリンの有効性は、少なくとも部分的には、BETの阻害および標的遺伝子転写の抑制によるものである。 BET阻害剤としてのニトロキソリンの発見は、BETファミリー関連疾患の治療のためのニトロキソリンおよびその誘導体の潜在的用途を示唆している。
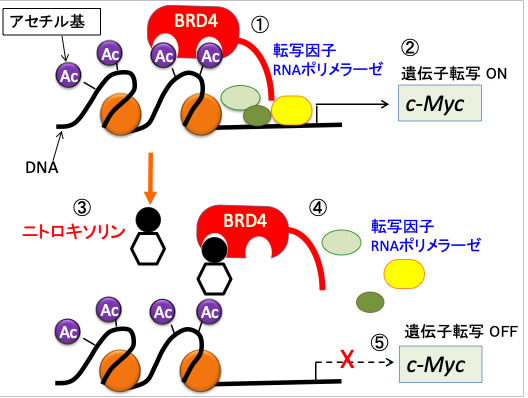
BET (bromodomain and extra-terminal)ファミリータンパク質は、ブロモドメインにおいてヒストンのアセチル化されたリシンを認識することにより、転写活性化因子として機能します。BRD4はこのようなBETファミリータンパク質の1つです。 BETファミリータンパク質のBRD4の阻害剤(=ニトロキソリン)はc-Mycの発現を抑制する作用があると言えます。
図:ヒストンのアセチル化されたリシンを認識するブロモドメインの繰り返し配列と特異的末端配列を持つBET(bromodomain and extra-terminal)ファミリータンパク質の一つのBRD4は、ヒストンのアセチル化リシンに結合し、転写因子やRNAポリメラーゼなどをリクルートして(①)、c-Myc遺伝子の転写を促進する(②)。ニトロキソリンはBRD4の第一ブロモドメインとアセチル化ヒストンの結合を阻害する(③)。その結果、転写因子やRNAポリメラーゼのリクルートが阻害され(④)、c-Myc遺伝子の転写を抑制する(⑤)。
ニトロキソリンの尿路感染症の治療に使う量は1日に500から750mgです。この尿路感染症に使用する服用量で十分な抗腫瘍効果が期待できることが動物実験の研究で報告されています。
がん治療に関するご質問やお問合せは、電話(03-5550-3552)かメール(info@f-gtc.or.jp)でお願いします。