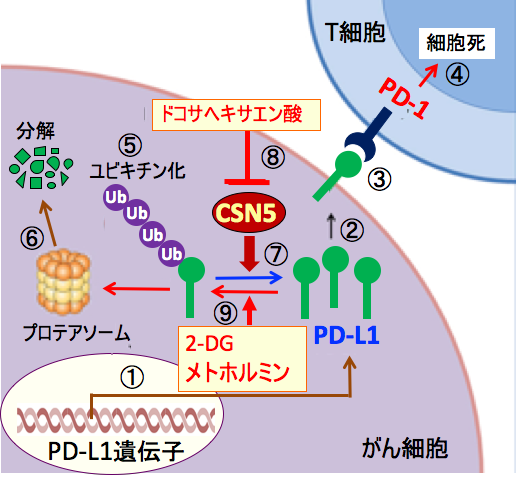
がん細胞のPD-L1を減らすドコサヘキサエン酸と2-デオキシ-D-グルコースとメトホルミン
【体にはがん細胞を排除する免疫監視機構が備わっている】
免疫系は自己と非自己を識別し、非自己を排除する生体防御システムです。 異物(非自己)を排除する免疫系は、自己の変異細胞であるがん細胞も排除して生体を防御しています。 免疫力の低下ががんの発生や進行を促進することは多くの証拠があります。 免疫抑制剤を使用している臓器移植患者や、HIV感染(エイズ)などによる免疫不全状態の患者はがんの発生率が高いことが知られています。
免疫力を高めてがんの消滅を目的とする「免疫療法」は、手術、化学療法、放射線療法に次ぐ第4のがん治療法として重要視されています。 溶連菌製剤のピシバニールや、カワラタケの菌糸体から見つかったタンパク結合多糖(ベータグルカンにタンパク質が結合)のクレスチンなど、免疫系を活性化する目的のがん治療薬もあります。
最近では、オプジーボなどの免疫チェックポイント阻害剤で、一部のがん患者において、がんが縮小したり消滅することが明らかになっています。 つまり、体に備わった免疫監視機構を十分に高めることができれば、がん細胞を死滅させ、がんを縮小したり、消滅させることもできるのです。
【抗原提示とT細胞の活性化】
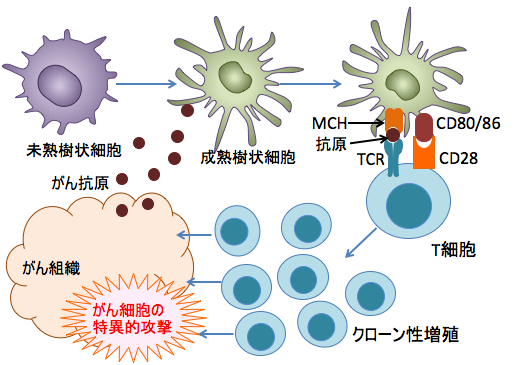
リンパ球のT細胞は、がん抗原で活性化されて初めて細胞傷害活性を持つようになります。すなわち、細胞傷害活性を持たないT細胞が抗原提示細胞(マクロファージや樹状細胞)から抗原ペプチド(がん抗原)を提示されて活性化してはじめてがん細胞に対して特異的な細胞傷害活性を持つ細胞傷害性T細胞(キラーT細胞)となり、がん細胞を攻撃するようになります。
病原微生物が侵入したり、何らかの原因で炎症が起こると、血管から顆粒球や単球などが遊走して来ます。このように炎症反応によって集まってきたり、あるいは組織に常在していた樹状細胞やマクロファージは、侵入した細菌やウイルス粒子、あるいは死滅した細胞の死骸や断片などを取り込み、リンパ液の流れに沿って所属リンパ節に移動します。
樹状細胞やマクロファージは取り込んだタンパク質を分解し、その結果産生されたペプチド(アミノ酸が数個から数十個つながったもの)をMHC(major histocompatibility complex:主要組織適合抗原複合体)分子の上に提示します。 活性化した樹状細胞はリンパ節で手当たりしだいにナイーブT細胞(まだ一度も活性化されたことのないT細胞)とくっつきあって、何かを確かめます。
ナイーブT細胞はその表面にT細胞抗原認識受容体(TCR)を持っています。樹状細胞の表面に提示されたMHC+抗原ペプチドとピタッとくっつく受容体(TCR)をもったナイーブT細胞と出会うと、そのT細胞を活性化します。 抗原を提示して活性化している樹状細胞にはCD80/86という補助刺激因子が発現しており、T細胞のCD28と結合し、刺激を送ります。 さらに、活性化した樹状細胞はサイトカインを放出しており、ナイーブT細胞はそれを浴びることになります。 このように、TCRを介するシグナルとCD28を介する補助刺激とサイトカインによる刺激を同時に受けたTリンパ球は初めて活性化し、TCRの特異性を保ったままで分裂・増殖して自らのクローンを増やします。
CD8陽性T細胞(キラーT細胞)は成熟し、細胞質内にパーフォリンやグランザイムなどを含んだ細胞傷害顆粒を持つエフェクター細胞になります。 エフェクター細胞はリンパ節を離れ、胸管を経て循環血液中へと流れ込み、血流に従って全身を巡ります。
図:がん細胞から放出されたがん抗原を未熟樹状細胞が取り込んで成熟して抗原を提示するとき、MHC(major histocompatibility complex:主要組織適合抗原複合体)分子にペプチド抗原を載せて細胞傷害性T細胞やヘルパーT細胞に提示する。このとき、MCH+ペプチド抗原にぴったり結合するTCR(T細胞受容体)を持つT細胞は、補助刺激因子(CD28とCD80/86など)や樹状細胞から放出されるサイトカインの働きで活性化され、がん抗原を認識するT細胞がクローン性増殖(clonal expansion)し、がん細胞を攻撃する。
【抗がん剤や放射線は抗腫瘍免疫を刺激する】
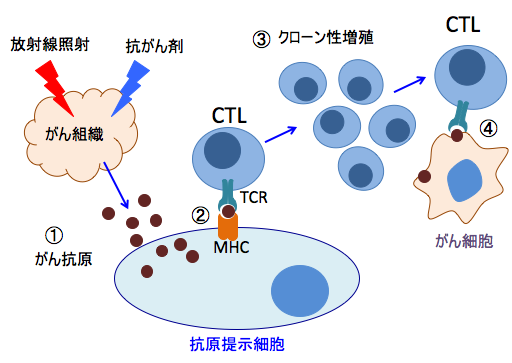
放射線治療が全身の抗腫瘍免疫の活性化の引き金になりうることが明らかになっています。 がん組織に放射線照射を行うと、がん細胞が死滅して細胞内成分が放出されるとこれらの成分が危険シグナルとなって自然免疫が活性化されます。同時に死滅したがん細胞からがん抗原が放出され、このがん抗原の情報を抗原提示細胞(樹状細胞やマクロファージ)が細胞傷害性T細胞(CTL)に提示してCTLは活性化され、獲得免疫が成立すると、生き残ったがん細胞を攻撃して排除しようとします。
同様に、抗がん剤治療も、分裂しているがん細胞を死滅させるだけでなく、この死滅した細胞から放出された細胞成分が自然免疫を刺激し、がん抗原が樹状細胞などの抗原提示細胞に認識されて、がん抗原特異的な抗腫瘍免疫を引き起こします。 抗がん剤治療と免疫チェックポイント阻害剤の併用が相乗効果を示し、奏功率の向上が報告されています。
図:放射線治療や抗がん剤治療でがん細胞が死滅するとがん抗原が放出される(①)。がん抗原は樹状細胞やマクロファージなどの抗原提示細胞に取込まれ、ペプチドに分解されて抗原ペプチドとして抗原提示細胞上のMHC(主要組織適合抗原複合体)に提示される(②)。MHCはがん抗原を介してCTL(細胞傷害性T細胞)上のTCR(T細胞受容体)と反応してCTLを活性化し、抗原提示を受けたがん抗原特異的なCTLはクローン性に増殖し(③)、がん抗原を持っているがん細胞を攻撃する(④)。
【細胞傷害性T細胞を抑制するPD-1とCTLA-4】
リンパ球の一種のT細胞は、病原菌やがん細胞を攻撃・排除する働きがあります。しかし、T細胞が暴走して正常な細胞を攻撃すると危険なので、いくつかのブレーキ装置が備わっています。これを「免疫チェックポイント」と呼びます。 がん細胞は、ときに巧みにこの免疫チェックポイントを悪用して、T細胞にブレーキをかけてT細胞からの攻撃を逃れようとするのです。
がん細胞によるブレーキがかからないようにする薬が免疫チェックポイント阻害薬です。
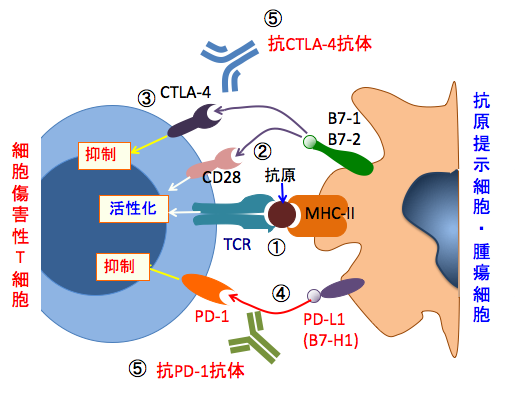
活性化した細胞傷害性T細胞にはPD-1やCTLA-4という受容体が発現します。PD-1はプログラム細胞死1(programmed death-1)、CTLA-4は細胞傷害性Tリンパ球抗原-4 (cytotoxic T-lymphocyte-associated protein 4)の略です。これらの受容体のリガンド(受容体に結合して作用する物質)となるPD-L1やB7(B7-1, B7-2)を抗原提示細胞が持つことによって細胞傷害性T細胞の働きを抑制しています。 つまり、PD-1受容体やCTLA-4受容体がリガンドによって刺激されると、T細胞の増殖が停止し細胞死を来すことになります。このようにして細胞傷害性T細胞の過剰な応答を制御しています。
細胞傷害性T細胞の働きを阻害するPD-L1やB7はがん細胞にも発現しています。つまり、がん細胞は免疫系の制御システムを利用して、がん組織内の細胞傷害性T細胞の働きを阻止しています。 PD-1受容体やCTLA-4受容体は細胞傷害性T細胞を死滅させるスイッチなようなものなので、これらのスイッチが入らないようにすれば、細胞傷害性T細胞は生き残ってがん細胞の攻撃力を高めることができます。 CTLA-4に対する抗体(ヒト型抗ヒトCTLA-4モノクローナル抗体)のイピリブマブ(ipilimumab: YERVOY)やヒト型抗PD-1モノクローナル抗体のニボルマブ(nivolumab商品名「オプジーボ(Opdivo)」)などがあります。このような免疫チェックポイント阻害剤を使用すると、がん細胞を攻撃する細胞傷害性T細胞の働きを高めることが可能になります。
図:抗原提示細胞上にはMHCクラスII(MHC-II)といわれる分子があり、抗原を介してT細胞上のTCR(T細胞受容体)と反応して細胞傷害性T細胞を活性化する(①)。T細胞上にはCD28とCTLA-4があり、CD28は恒常的に発現し、抗原提示細胞からのB7-1やB7-2というリガンドによってT細胞活性化に作用する(②)。一方、CTLA-4はT細胞活性化にともなって発現が誘導され、B7-1やB7-2によって刺激されるとT細胞を抑制する(③)。CTLA-4はCD28よりもB7に対する親和性が強いので、活性化したT細胞の過剰な応答を抑制する。同様に、PD-1(Programmed death-1)は抗原提示細胞のPD-L1(別名B7-H1)と結合することによって抑制型の免疫調節シグナルを活性化させる(④)。がん細胞もB7-1やB7-2やPD-L1が発現しており、細胞傷害性T細胞の働きを抑制している。T細胞のCTLA-4とPD-1の働きを特異抗体で阻害すると、がん細胞に対する細胞傷害性T細胞の働きを高めることができる(⑤)。
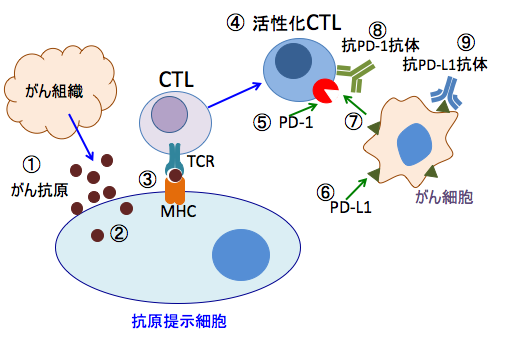
がん細胞を非自己と認識して、それを攻撃するためにT細胞は活性化しますが、PD-1リガンド(PD-L1)を持ったがん細胞と接触すると、CTL上のPD-1とリガンド(PD-L1)が結合することにより、免疫シグナルは抑制され、T細胞はがん細胞を攻撃できなくなってしまいます。これがT細胞を活性化するだけの従来の免疫療法に限界があった理由です。
活性化したCTL(細胞傷害性T細胞)をがん組織に送っても、がん細胞を攻撃しようと近づくとPD-L1によって自身のPD-1のスイッチが入って死滅するからです。
T細胞やNK細胞を活性化すると同時に、T細胞上の受容体(PD-1やCTLA-4)の働きを阻止したり、がん細胞に発現しているPD-1のリガンド(PDL-1)の発現量を減らす方法、PD-1とPD-L1の結合を阻害する方法も有効です。
図:がん細胞から放出されたがん抗原(①)は、樹状細胞やマクロファージなどの抗原提示細胞に取込まれ(②)、ペプチドに分解されて抗原ペプチドとして抗原提示細胞上のMHC(major histocompatibility complex:主要組織適合抗原複合体)に提示される(③)。MHCはがん抗原を介してCTL(細胞傷害性T細胞)上のTCR(T細胞受容体)と反応してCTLを活性化する(④)。CTLは活性化されるとPD-1(Programmed death-1)が発現する(⑤)。がん細胞にはPD-1のリガンドであるPD-L1が発現している(⑥)。PD-1とPD-L1が結合するとCTLは増殖が抑制される(⑦)。PD-1とPD-L1の結合を抗PD-1抗体(⑧)や抗PD-L1抗体(⑨)で阻止するとCTLの抗腫瘍活性を高めることができる。
【PD-L1のユビキチン・プロテアソーム系による分解】
プログラム細胞死タンパク質 1 (PD-1) や細胞傷害性 T リンパ球関連タンパク質 4 (CTLA-4) などの免疫チェックポイントを標的とする抗体の先駆的な成功は、がん治療の展望を変えました。 オプジーボなどの抗体ベースの治療から、経口投与可能で低分子の物質による免疫チェックポイント阻害剤の開発に注目が集まっています。
PD-L1はユビキチン・プロテアソーム系で分解されるため、PD-L1の分解を促進して、PD-1/PD-L1経路による細胞傷害性Tリンパ球の抑制を阻止する方法もターゲットの一つになっています。
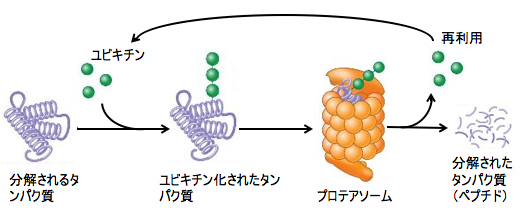
ユビキチン・プロテアソーム系はタンパク質に付加されたユビキチン鎖をプロテアソームが認識し,ATP依存的で迅速かつ不可逆に標的タンパク質を分解するシステムです。 ユビキチン(Ubiquitin)は,アミノ酸76残基からなり,酵母からヒトまであらゆる真核細胞に存在する進化的に保存されたタンパク質です。 ユビキチンは不要なタンパク質、たとえば折り畳み不全などの出来損なったタンパク質や古くなったタンパク質に複数個付加(ポリユビキチン化)されることで、タンパク質分解のシグナルとして働きます。つまり、「このタンパク質を分解してくれ」という目印になります。
標的タンパク質へのユビキチン付加反応はユビキチン活性化酵素(E1)、ユビキチン結合酵素(E2)、およびユビキチンをE2から特定の基質に送達するユビキチンリガーゼ(E3)によって行われます。 ユビキチン自体はあくまで目印なので、分解を行うのは他の物質です。ユビキチンが結合した不要たんぱく質をシュレッダーのように分解する酵素をプロテアソームといいます。 プロテアソームは真核生物のATP依存性プロテアーゼ複合体で、分解目印として働くユビキチンが結合したたんぱく質を選択的に壊す複雑な細胞内装置です。
図:分解されるタンパク質はユビキチンが複数個結合し、ユビキチンが結合したタンパク質をプロテアソームが認識して、タンパク質を分解する。
PD-L1はユビキチン化と分解を受けますが、がん細胞は複数の経路によってこのプロセスを阻害する能力を示し、腫瘍の免疫抑制をもたらします。つまり、がん細胞はTリンパ球からの攻撃を防ぐ目的でPL-D1の分解を阻止しているのです。したがって、がん細胞のPD-L1のユビキチン化を促進し、プロテアソームでの分解を促進する方法はがん細胞の増殖を抑える効果が期待できます。
【ドコサヘキサエン酸はPD-L1の分解を促進する】
オメガ3系多価不飽和脂肪酸のドコサヘキサンエン酸(DHA)がPD-L1の分解を促進することが報告されています。以下のような論文があります。
Docosahexaenoic acid reverses PD-L1-mediated immune suppression by accelerating its ubiquitin-proteasome degradation.(ドコサヘキサエン酸は、そのユビキチン-プロテアソーム分解を加速することにより、PD-L1 を介した免疫抑制を逆転させる)J Nutr Biochem. 2022 Oct 27;112:109186.
【要旨の抜粋】
この研究では、ω-3 多価不飽和脂肪酸のドコサヘキサエン酸 (DHA) が in vitro と in vivo の両方でがん細胞の PD-L1 の発現を低下させることを初めて実証した。
DHA はユビキチン-プロテアソームによるPD-L1分解を促進してPD-L1 発現の減少をもたらし、PD-L1 を介した免疫抑制を逆転させ、腫瘍増殖を阻害する。 さらに、DHA はがん細胞の脂肪酸合成酵素の発現を有意に減少させ、これはパルミトイルトランスフェラーゼ DHHC5 を阻害し、CSN5 依存性 PD-L1 分解を促進した。
今回の発見により、DHA の抗がん作用に関与する新しいメカニズムが明らかになり、DHA ががんの治療と予防のための新しい免疫増強剤として開発される有望な可能性を秘めていることが示唆された。上記の論文で、「CSN5 依存性 PD-L1 分解」のCSN5は「constitutive photomorphogenic-9 signalosome 5」の略です。CSN5 は、DNA 修復の調節、シグナル伝達の調節、および細胞増殖の制御に関与する、進化的に保存された多機能タンパク質です。CSN5 は、細胞内の P27、P53、およびサイクリン E を機能的に不活性化することにより、細胞の生存に寄与する発がん性タンパク質として機能します。 CSN5 の最もよく理解されている機能は、脱ユビキチン化活性によるユビキチンを介したタンパク質分解の調節です。例えば、CSN5はサバイビン(survivin)とsnailのユビキチン化と分解をブロックし、がん細胞の浸潤と移動を促進します。
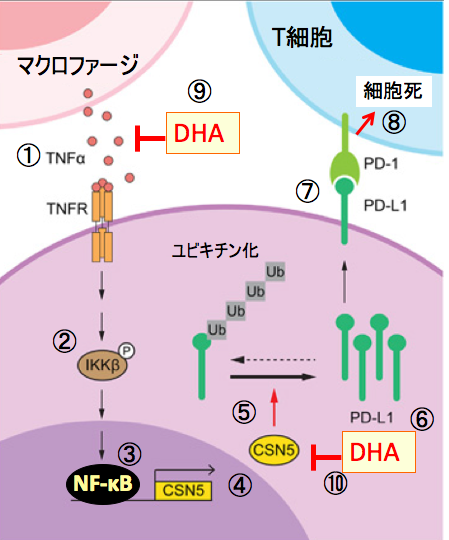
腫瘍壊死因子-α(TNF-α) 誘導の CSN5 発現は、がん細胞の PD-L1 安定化をもたらし、免疫監視から逃れるメカニズムの一つになっています。CSN5 は複数のヒトがんで発現が亢進しており、予後不良と関連しています。 ドコサヘキサエン酸は抗炎症作用があり、炎症性サイトカイン(TNF-α、IL-6など)の産生を抑制し、NF-κBの活性化を抑制します。 ドコサヘキサエン酸はCSN5活性の阻害と、抗炎症作用によるNF-κBの活性化の抑制という2つのメカニズムでがん細胞のPD-L1の発現を抑制します。
図:がん組織内ではマクロファージからTNF-αが産生され、がん細胞のTNF受容体(TNFR)を刺激し(①)、Iκκβをリン酸化して活性化し(②)、NF-κBの転写活性を行進し(③)、CSN5を産生する(④)。CSN5はPD-L1のユビキチン化を阻害して分解を阻害し(⑤)、PD-L1の量を増やす(⑥)。がん細胞のPD-L1はT細胞のPD-1と結合し(⑦)、T細胞の細胞死を誘導する(⑧)。ドコサヘキサエン酸(DHA)はマクロファージからのTNF-αの産生を阻止してNF-κBの転写活性を抑制する(⑨)。さらに、DHAはCSN5の活性を阻害する(⑩)。これらの作用によってDHAはがん細胞のPD-L1の発現を低下させる。
【PD-L1のグリコシル化がターゲットになる】
タンパク質は遺伝子によって決められた配列によってアミノ酸が結合して作られます。タンパク質が作られるとき、まず遺伝子(DNA)からメッセンジャーRNAが転写されます。このメッセンジャーRNAからタンパク質が合成される過程を「翻訳」と言います。メッセンジャーRNAからポリペプチドへの翻訳はリボソームで行われます。翻訳後のポリペプチド鎖は小胞体で3次元的に折り畳まれます。
できたタンパク質はさらにリン酸やアセチル基や糖鎖などが結合して、タンパク質の活性や働きが変化します。このようなタンパク質の修飾を翻訳後修飾と言います。このような翻訳後修飾によってタンパク質の働きが制御されています。
PD-L1のグリコシル化(糖鎖結合)を阻害すると、PD-L1の分解が促進され、PD-1/PD-L1軸による免疫抑制が阻止できることが報告されています。 以下のような論文が最近報告されています。
B7 family protein glycosylation: Promising novel targets in tumor treatment.(B7ファミリーのタンパク質グリコシル化:腫瘍治療における有望な新規標的)Front Immunol . 2022 Dec 6;13:1088560.
B7ファミリーは免疫グロブリン様ドメインがある膜貫通型の糖タンパクです。B7ファミリーの分子は共刺激または共阻害能があり、主にCD28、CTLA-4、PD-1などのCD28ファミリーメンバーと結合し、共刺激シグナルと共抑制シグナルをそれぞれ送信することにより、T 細胞応答を誘導または阻害します。
B7 ファミリー メンバーである PD-L1、PD-L2、B7-H3、および B7-H4 のグリコシル化をブロックすると、これらの免疫チェックポイント・タンパク質の自己安定性と受容体結合が阻害され、その結果、免疫チェックポイント阻害作用を発揮し、抗腫瘍免疫を増強し、腫瘍の増殖を抑制します。したがって、PD-L1などのB7ファミリーのタンパク質のグリコシル化の調節は、腫瘍の免疫抑制を緩和するための重要な鍵となる可能性を指摘しています。
【2-デオキシ-D-グルコースはPD-L1の分解を促進する】
Deglycosylation of PD-L1 by 2-deoxyglucose reverses PARP inhibitor-induced immunosuppression in triple-negative breast cancer. (2-デオキシグルコースによるPD-L1の脱グリコシル化は、トリプルネガティブ乳がんにおけるPARP阻害剤誘発性免疫抑制を逆転させる)Am J Cancer Res. 2018; 8(9): 1837–1846.
がん細胞のPD-L1は免疫細胞の働きを阻害します。抗がん剤のPARP阻害剤はPD-L1の発現を亢進して抗腫瘍免疫を抑制します。2-DGはPARP阻害剤によって誘導されるPD-L1発現亢進に対して、PD-L1のグリコシル化(糖鎖結合)を阻害してPD-L1の作用を阻害し、抗腫瘍免疫を回復させることを報告しています。以下のような報告もあります
Saccharide analog, 2-deoxy-d-glucose enhances 4-1BB-mediated antitumor immunity via PD-L1 deglycosylation.(糖類似体の2-デオキシ-d-グルコースは、PD-L1 脱グリコシル化を介して 4-1BB を介した抗腫瘍免疫を増強する)Mol Carcinog. 2020 Jul;59(7):691-700.
【要旨の抜粋】
プログラム細胞死タンパク質 1 (PD-1)/プログラム死リガンド 1 (PD-L1) 遮断療法は、トリプルネガティブ乳がん患者で 10% から 20% の応答率を示す。 私たちの以前の研究は、PD-L1 タンパク質が トリプルネガティブ乳がんで高度にグリコシル化されており、グリコシル化(糖鎖の結合)が PD-L1 タンパク質の安定性と免疫抑制機能に重要な役割を果たしていることを示している。この報告では、糖類似体である 2-デオキシ-D-グルコース (2-DG) が、EGFR 阻害剤であるゲフィチニブと組み合わせることにより、PD-L1 のグリコシル化とその免疫抑制機能を阻害することを発見した。
興味深いことに、2-DG/ゲフィチニブによる PD-L1 の脱グリコシル化は、PD-L1 タンパク質の発現レベルと PD-1 との結合を減少させた。
しかし、2-DG/ゲフィチニブによる 4-1BB の発現と 4-1BBL との結合に有意な減少は認めなかった。 さらに、2-DG/ゲフィチニブと 4-1BB 抗体の併用治療が TNBC 同系マウスモデルで抗腫瘍免疫を増強することを実証した。
まとめると、我々の結果は、トリプルネガティブ乳がんにおける PD-L1 脱グリコシル化と 4-1BB 刺激によって抗腫瘍免疫を強化する新しい免疫療法戦略を示唆している。
4-1BB は、腫瘍組織に浸潤した細胞傷害性T細胞に選択的に発現している共刺激分子で、4-1BB を標的とした刺激抗体を用いることにより、腫瘍組織内の腫瘍特異的な細胞傷害性T細胞を選択的に活性化し、抗腫瘍効果を発揮します。 その作用機序から予想されるようにPD-1/PD-L1経路を阻害することは4-1BBを刺激する抗体療法の抗腫瘍効果を高めることができます。
2-デオキシ-D-グルコースはPD-L1のグリコシル化を阻害することによってPD-L1の免疫抑制効果は阻止できるという報告です。
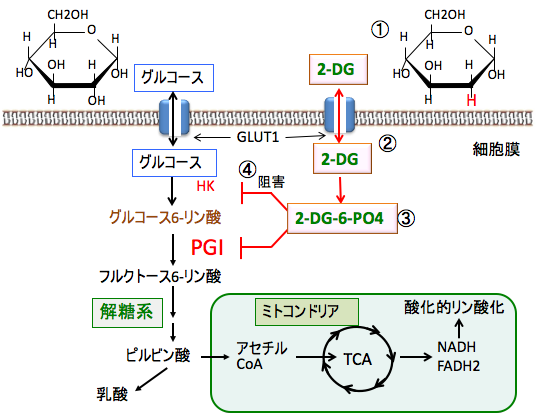
2-デオキシ-D-グルコース(2-Deoxy-D-Glucose:2-DG)はグルコース(ブドウ糖)の2位のOHがHに変わっているグルコース類縁物質です。 2-DGはグルコースと同じトランスポーター(輸送担体)で取り込まれます。細胞内では、ヘキソキナーゼによってリン酸化されて、2-デオキシグルコース-6リン酸(2-DG-6リン酸)に変換されますが、この2-DG-6リン酸は解糖系の先の代謝系には進めない(ヘキソキナーゼの先の解糖系酵素で代謝できない)ので、ATP産生量が減ります。
さらに、蓄積した2-DG-6リン酸はヘキソキナーゼを阻害する作用もあるので、正常なグルコースの代謝も阻害されます。 がん細胞はグルコースの取り込みが亢進しており、2-DGの取り込みも増えているので、がん細胞の解糖系を阻害する効果でがん治療に使われています。
2-デオキシ-D-グルコース(2-DG)は小胞体ストレスを高めてがん細胞を死滅させる作用が報告されています。さらに、2−デオキシ-D-グルコースは抗がん剤や放射線の免疫原性細胞死を増強することが報告されています。
図:2-デオキシ-D-グルコース(2-DG)はグルコース(ブドウ糖)の2位のOHがHに変わっているグルコース類縁物質(①)で、グルコースと同様にグルコーストランスポーター(GLUT1)によって細胞内に取り込まれる(②)。細胞内のヘキソキナーゼで2-DG-6リン酸(2-DG-6-PO4)になるが、それから先の解糖系酵素では代謝できないので細胞内に蓄積する(③)。蓄積した2-DG-6リン酸はヘキソキナーゼ(HK)とホスホグルコースイソメラーゼ(PGI)をフィードバック的に阻害するので、グルコースの解糖系での代謝を阻害する(④)。
小胞体(Endoplasmic reticulum)は、細胞内における分泌・膜タンパク質の品質管理において大切な小器官です。 2-DGは解糖系を阻害する以外に、タンパク質に糖鎖が着くN-グリコシル化の過程を阻害するので、糖タンパク質の生成を阻害します。
グリコシル化というのはタンパク質に糖類が付加する反応で、小胞体で行われて、正常に糖が付加したタンパク質はゴルジ体に運ばれます。 糖鎖異常の糖タンパク質は、折り畳みが不完全な異常タンパク質になり、小胞体に蓄積して小胞体ストレスを引き起こし、細胞死の原因にもなります。 つまり、2-デオキシ-D-グルコース(2-DG)はタンパク質のN-グリコシル化(N-glycosylation)を阻害するので小胞体ストレスを高めて免疫原性細胞死を増強する作用があります。
抗がん剤でがん細胞を死滅させるときに2−DGを投与しておくと、死滅したがん細胞は免疫原性が高くなるので、がん抗原特異的な抗腫瘍免疫を誘導でき、延命効果を高めることができます。 さらにPD-1/PD-L1軸による免疫抑制を阻害するので、抗腫瘍免疫を高める効果を増強します。
【メトホルミンはPD-L1の分解を促進する】
糖尿病治療薬のメトホルミンがPD-L1の分解を促進することは多くの報告があります。例えば以下のような報告があります。
Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1.(メトホルミンは小胞体関連の PD-L1 分解を介して抗腫瘍免疫を促進する)Mol Cell. 2018 Aug 16;71(4):606-620.e7.
【要旨】
メトホルミンは、抗腫瘍活性を有し、高い細胞傷害性 T リンパ球 (CTL) 免疫監視を維持することが報告されている。 ここでは、メトホルミンがプログラム死リガンド-1 (PD-L1) の安定性と膜局在を低下させることにより、CTL 活性を増加させることを示す。
さらに、メトホルミンによって活性化された AMP 活性化プロテインキナーゼ (AMPK) が PD-L1 の S195 を直接リン酸化することを発見した。 S195 リン酸化は異常な PD-L1 グリコシル化を誘導し、小胞体への蓄積と小胞体でのPD-L1タンパク質の分解を引き起こす。
メトホルミン治療を受けた乳がん患者の腫瘍組織は、AMPK 活性化により PD-L1 レベルの低下を示す。
メトホルミンによる PD-L1 の阻害シグナルの遮断は、がん細胞に対する 細胞傷害性 T リンパ球 (CTL) 活性を高める。 私たちの調査結果は、小胞体におけるPD-L1タンパク質の分解を介したPD-L1発現の新しい調節メカニズムを特定し、メトホルミン-CTLA4遮断の組み合わせが免疫療法の有効性を高める可能性があることを示唆している。
Metformin suppresses cancer cell growth in endometrial carcinoma by inhibiting PD-L1.(メトホルミンは PD-L1 を阻害することにより、子宮内膜がんのがん細胞増殖を抑制する)Eur J Pharmacol. 2019 Sep 15;859:172541.
【要旨の抜粋】
メトホルミンはがん患者の生存予後を臨床的に改善することがわかっている。 子宮内膜がん細胞株 Ishikawa および RL95-2を使用し、メトホルミンの作用を検討した。
子宮内膜がん細胞に対して、メトホルミン治療がPD-L1タンパク質の発現レベルを低下させることが認められた。 メトホルミン処理は、子宮内膜がん細胞に対する T細胞の抗腫瘍効果を有意に亢進した。 我々は、メトホルミンによる PD-L1 の阻害が AMPKに依存していること、およびメトホルミンが PD-L1 タンパク質とAMPK タンパク質の直接結合を促進することを実証した。 糖尿病治療に使用される従来の薬であるメトホルミンが、子宮内膜がんの抗腫瘍活性を保持することを確認した。
メトホルミンの抗腫瘍効果は、PD-L1 発現の阻害と AMPK シグナル伝達タンパク質の活性化に関連しており、メトホルミンの抗腫瘍特性に新しいメカニズムを提供する。Dual Effect of Combined Metformin and 2-Deoxy-D-Glucose Treatment on Mitochondrial Biogenesis and PD-L1 Expression in Triple-Negative Breast Cancer Cells(トリプルネガティブ乳がん細胞におけるミトコンドリア新生と PD-L1 発現に対するメトホルミンと 2-デオキシ-D-グルコースの併用治療の二重効果)Cancers (Basel) . 2022 Mar 5;14(5):1343.
【要旨】
メトホルミンと 2-デオキシ-D-グルコース (2DG) は、がん細胞の増殖抑制や PD-L1 発現抑制など、代謝および免疫調節に関連する複数のメカニズムで抗がん効果をする。
本研究では、in vitro で トリプルネガティブ乳がん細胞のミトコンドリアに対するメトホルミン、2DG、およびそれらの組み合わせ (メトホルミン + 2DG) の効果を調査した。
メトホルミン + 2DG は、トリプルネガティブ乳がん細胞のミトコンドリア質量を増加させた。 これは、形態学的に正常なミトコンドリアの数の増加ではなく、ミトコンドリアのサイズの増加に関連しており、ミトファジーの抑制ではなく、ミトコンドリア新生の誘導によって引き起こされた。
2DG およびメトホルミン + 2DG は、タンパク質の N-グリコシル化を阻害することにより、小胞体ストレス応答を強く誘導した。 適切なエネルギーストレスとともに、これはミトコンドリア拡大の可能性のあるトリガーの1つであった。
2DG またはメトホルミン + 2DG による N-グリコシル化の抑制はPD-L1 脱グリコシル化も引き起こし、MDA-MB-231 細胞におけるPD-L1の発現を減少させた。 PD-L1 は低グルコースで増加し、両方の薬剤で正常化された。 2DG およびメトホルミン + 2DG は、Jurkat 細胞の PD-1 発現を減少させたが、サイトカイン分泌はほとんど維持されていた。 したがって、メトホルミンと 2DG は、トリプルネガティブ乳がんの抗腫瘍免疫を改善するための補助療法として使用できる可能性がある。以上のような研究結果から、ドコサヘキサエン酸、2-デオキシ-D-グルコース、メトホルミンの組み合わせは、がん細胞に対する増殖抑制・アポトーシス誘導と、抗腫瘍免疫を高める効果によって、がん治療の補助療法として使用できる。
図:がん細胞はPD-L1の発現(①)が亢進しており、産生されたPD-L1は細胞表面に移行し(②)、T細胞のPD-1と結合して(③)、活性化した細胞傷害性T細胞の細胞死を誘導する(④)。PD-L1はユビキチン化されて(⑤)、プロテアソームで分解されている(⑥)。COP9シグナルソーム 5 (CSN5) はPD-L1に結合したユビキチンを外す作用があり、PD-L1のプロテアソームでの分解を阻止する(⑦)。ドコサヘキサエン酸はCSN5の脱ユビキチン化活性を阻害してPD-L1の分解を促進する(⑧)。2-デオキシ-D-グルコース(2-DG)とメトホルミンはPD-L1の糖鎖の異常を引き起こしてPD-L1のユビキチン化とプロテアソームでの分解を促進する(⑨)。